Overview
We seek to quantify the transmission dynamics of bacterial and viral infectious diseases using pathogen genome sequence data, with the ultimate goal of informing public health interventions to reduce the disease burden. Methodologically, we use phylodynamic methods to infer transmission dynamics from pathogen genome sequence data. We are particularly interested in developing methods to infer transmission dynamics from multiple data sources, such as pathogen genome sequences, epidemiological data, and spatial data. We apply these methods to study the transmission dynamics of influenza viruses, SARS-CoV-2, and bacterial pathogens.
An incomplete list of our research interests includes:
Local Phylodynamics to Inform Public Health Interventions
Phylodynamics is typically used to infer transmission dynamics of infectious diseases at a global scale. While global spread initiates novel outbreaks, the majority of cases result from local transmission, such as within households, schools, workplaces, public transportation, etc. We aim to develop computational approaches to infer local transmission dynamics of infectious diseases jointly from multiple data sources. Using this, we will investigate how we can utilize different sources of data collected as part of routine infectious diseases surveillance to quantify i) the relative disease burden in different neighborhoods and ii) the rates of transmission between different neighborhoods. Our goal is that a quantitative understanding of the local transmission dynamics will help to inform prevention strategies.
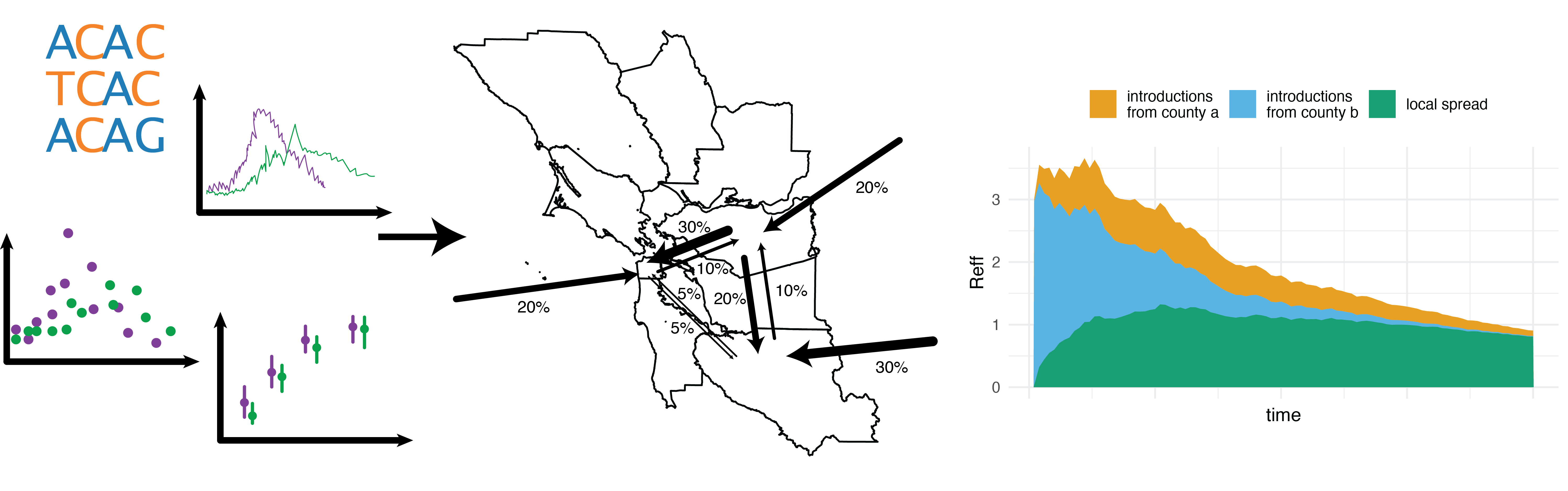
Relevant Publications:
- Viral genomes reveal patterns of the SARS-CoV-2 outbreak in Washington State
- Local-Scale phylodynamics reveal differential community impact of SARS-CoV-2 in metropolitan US county
- Characterising the epidemic spread of Influenza A/H3N2 within a city through phylogenetics
Genomic Epidemiology to Quantify the Spread of Bacterial Pathogens
Antibiotics have dramatically reduced the burden of bacterial pathogens on human health. Antimicrobial resistance threatens these achievements. Antimicrobial resistance can be acquired de novo or through horizontal gene transfer. Resistance genes can be spread, for example, between hospital and community settings or between humans and domesticated animals. How exactly this spread unfolds is unclear, and quantifying the relative contributions of different places or host types to the spread of antibiotic resistance is crucial to design efficient interventions to reduce the spread of resistance. Our goal is to develop phylodynamics approaches to quantify the spread of antibiotic resistance genes between different host types and geographic areas.
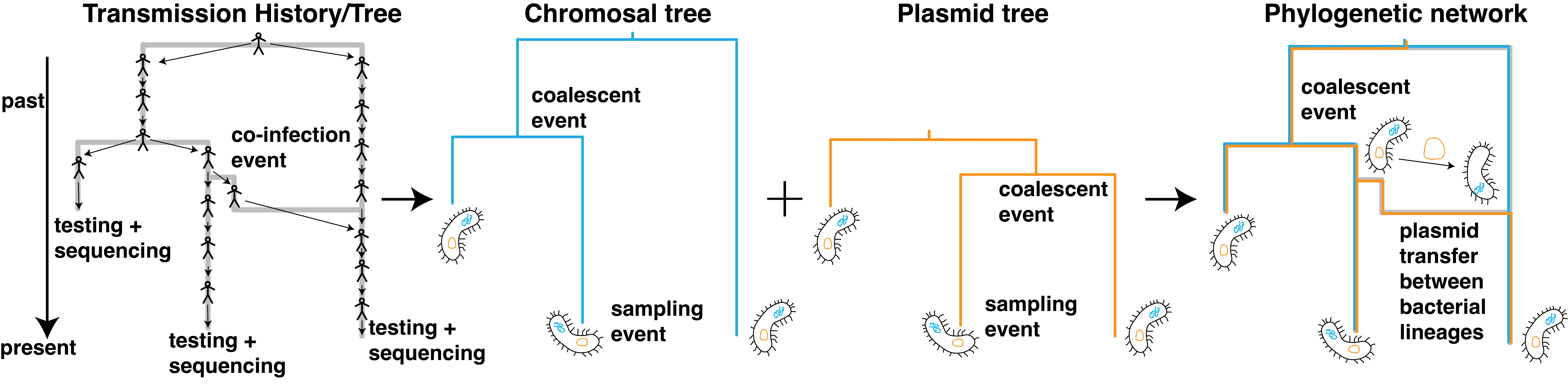
Relevant Publications:
- Tracking the horizontal transfer of plasmids in Shigella sonnei and Shigella flexneri using phylogenetics
- Phylodynamics Uncovers the Transmission of Antibiotic-Resistant Escherichia coli between Canines and Humans in an Urban Environment
How Recombination Shapes the Evolution and Transmission of Viruses
Recombination allows viruses to recombine genomic information from different ancestries. Different modes of recombination exist in RNA viruses such as reassortment in influenza viruses or template switching in coronaviruses. We seek to elucidate the role of recombination in the evolution and transmission of influenza and corona viruses. To do so, we use phylogenetic network approaches that reconstruct the ancestral history of viruses, identifies past recombination events, and provides insights in the spread and evolution of theses viruses. In particular, we are interested in the role reassortment in host switching of influenza viruses, and the how recombination impacts the fitness of endemic influenza and coronaviruses.
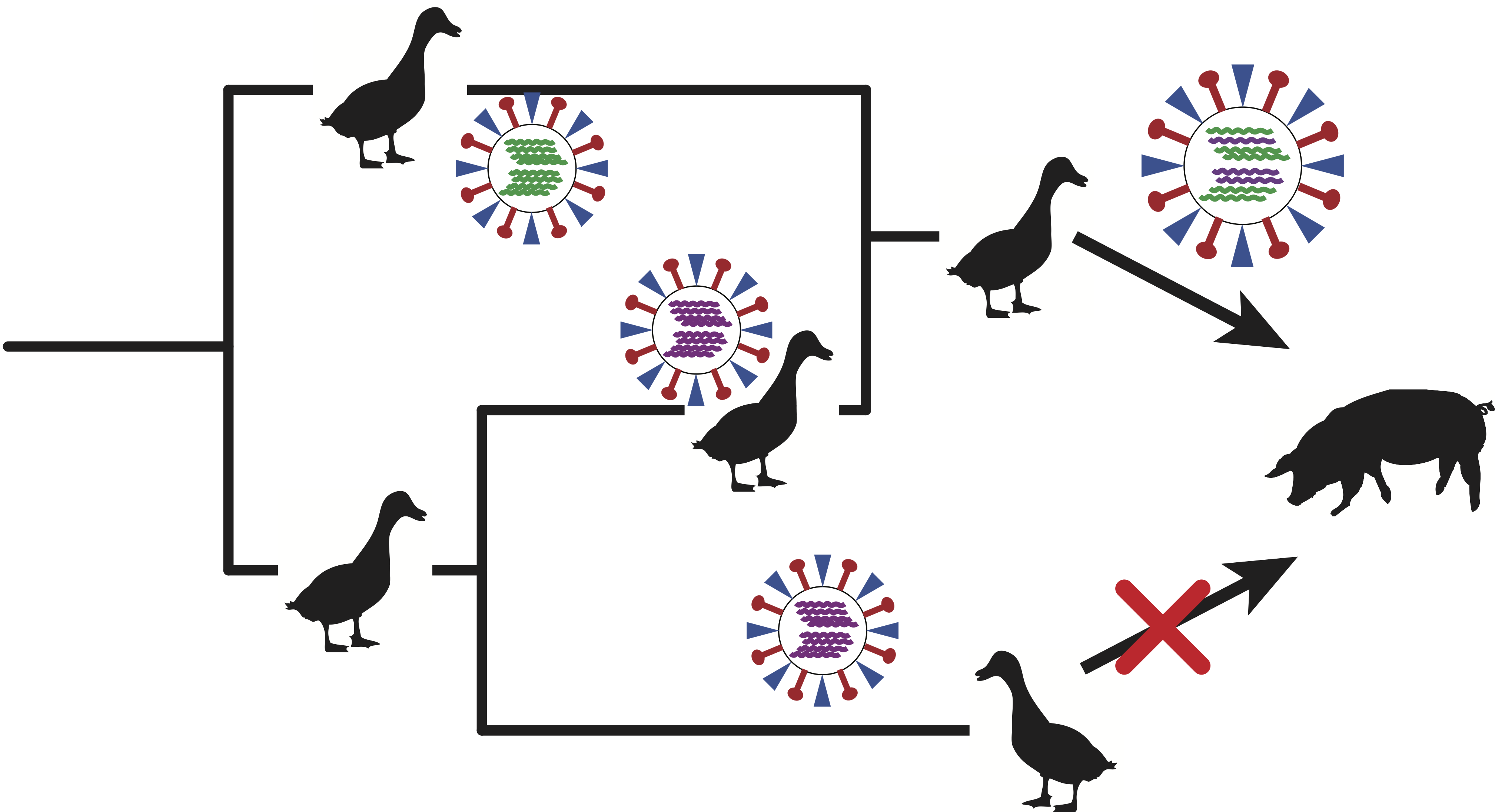
Relevant Publications: